incorporating optical mapping to genomic analysis
Even with significantly high coverage and various insert sizes, genome assembly and structural variation detection are tenuous computational processes using short read data alone due to repetitive regions in the genome. Thus, there is active interest in generating diverse types of data—to be used alone or in concert with short read data—in order to overcome the limitations of short read data and produce higher quality results. Optical maps, which are ordered genome-wide high-resolution restriction maps that specify the positions of occurrence of one or more short nucleotide sequences, are one such type of data.
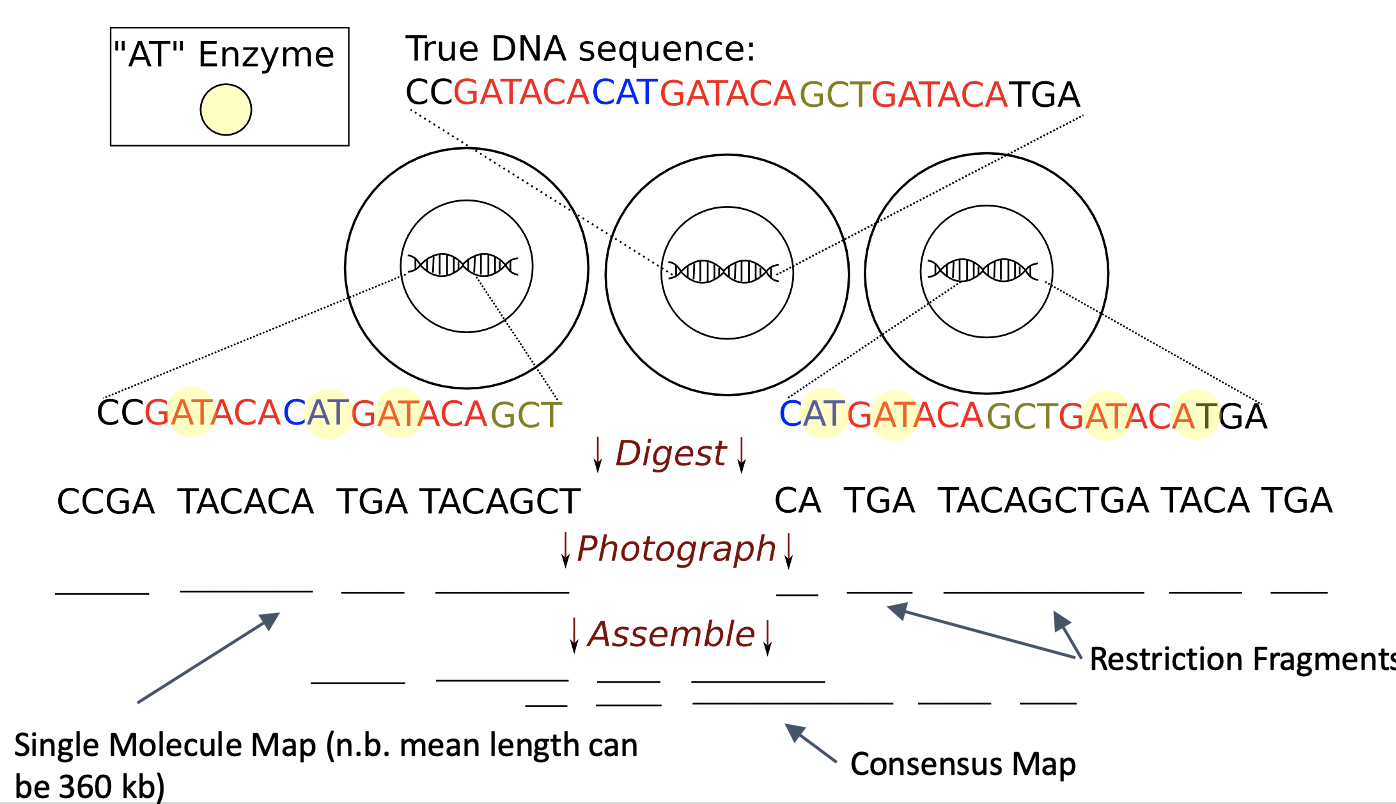
Optical mapping is a system for constructing ordered genome-wide high-resolution restriction maps, which can complement the information provided by paired-end sequence data. The system works as follows: first an ensemble of DNA molecules is adhered to a charged glass plate and elongated, enzymes are used to cleave them into fragments at loci where the enzymes’ recognition sequence occurs, these fragments are highlighted with fluorescent dye and imaged under a microscope, and finally, in order to produce a molecular map these images are analyzed to estimate the fragment sizes. My lab has developed several algorithms to analyze optical mapping data, alone or with sequence data.
Efficient Indexed Alignment of Contigs to Optical Maps (WABI 2014, WABI 2018, AMB 2019)
We presented the first index-based method for aligning assembled contigs to an optical map (WABI 2014) and thus, made a significant contribution. We refer to this method as Twin and demonstrate that by using a suitably-constructed FM-Index data structure. built on the optical map, the alignments between contigs and optical maps can be computed in time that is orders of magnitude than competing methods.
Disentangling De Bruijn Graphs Via Optical Maps (WABI 2017, WABI 2020, Algorithms in Molecular Biology)
While long reads have been shown to increase the quality of draft genomes in repetitive regions, fundamental computational challenges remain in overcoming their high error rate and assembling them efficiently. We show that the de Bruijn graph built on the long reads can be efficiently and substantially disentangled using optical mapping data as auxiliary information. Fundamental to our approach is the use of the positional de Bruijn graph and a succinct data structure for constructing and traversing this graph. Our experimental results show that over 97.7% of directed cycles have been removed from the resulting positional de Bruijn graph as compared to its non-positional counterpart. Our results thus indicate that disentangling the de Bruijn graph using positional information is a promising direction for developing a simple and efficient assembly algorithm for long reads.
This research is funded by the National Science Foundation (NSF III 1618814)
